肿瘤科
肿瘤分为良性肿瘤和恶性肿瘤。如脂肪瘤、子宫肌瘤、甲状腺腺瘤、部分胸腺瘤、畸胎瘤等良性肿瘤。恶性又称癌,按恶性细胞的来源可分为两大类,起源于上皮组织的称为癌,如肺癌、胃癌、胰腺癌等;起源于间叶组织的称肉瘤,如软组织和骨肉瘤、腹腔肉瘤等。
查看详细>>医学心理科
医学心理科主要的治疗手段包括药物治疗、暗示治疗、心理咨询与治疗、松弛治疗、音乐治疗、心理评估。主要服务:对各种身心疾病包括抑郁症、焦虑症、疑病症、强迫症、创伤后应激障碍、进食障碍、睡眠障碍、各种不明原因躯体不适以及各种躯体疾病伴发的情绪和睡眠等问题提供系统的药物治疗和专业的心理咨询及心理治疗。 诊疗范围: 行为问题(注意缺陷多动障碍、品行问题、对抗行为、电脑成瘾行为等)、情绪障碍(离别焦虑症、学校恐怖症、抑郁症、强迫症等)、发育性障碍(孤独症、精神发育迟滞等),以及其他心理问题
查看详细>>康复医学科
康复科引进的德国Woodway下肢康复机器人,统合了康复医学、生物力学、机械学、机械力学、电子学、材料学、计算机科学,是目前国内上较先进的自动化医疗康复设备之一。 德国Woodway下肢康复机器人是由步态训练器、woodway跑台系统、减重系统、固定绑带配件、操控系统,通过自主或被动康复训练,可重塑、纠正患者的步行方法;训练患者的行走能力;增强患者的血液循环和心肺功能;重塑患者运动神经;增强患者肌力和耐力;增加患者
查看详细>>外科
我院外科汇聚了资深的专家,成熟的技术,高端的设备,优质的服务等软硬件设施,主要以外科脉管畸形临床难题为重点,探索其先进治疗手段。专业治疗鲜红斑痣、静脉畸形、动静脉畸形、淋巴管畸形等常见而疑难的血管性疾病。
查看详细>>检验科
检验科是一个集医疗、教学于一体的现代化实验室,拥有先进的检测设备,健全的规章制度,较高素质的技术团队,较齐全的检测项目和较先进的检测技术。 目前,检验科设有采血室、血常规室、体液室等工作室,各实验室分工细密,人员各司其职,检测结果及时、准确。
查看详细>>
卢胜西
外科(血管瘤/血管畸形)
从事鲜红斑痣、婴幼儿血管瘤与脉管畸形研究和临床工作多年,对各类型血管瘤如浅表血管瘤、深浅混合型血管瘤、卡梅综合症、KTS综合症等疑难疾病治疗经验丰富。擅长动静脉畸形、淋巴管畸形、静脉畸形的诊断及微创治疗,对微创治疗后辅助手术方法修复复杂血管畸形颇有心得...
卢胜西
副主任医师
从事鲜红斑痣、婴幼儿血管瘤与脉管畸形研究和临床工作多年,对各类型血管瘤如浅表血管瘤、深浅混合型血管瘤、卡梅综合症、KTS综合症等疑难疾病治疗经验丰富。擅长动静脉畸形、淋巴管畸形、静脉畸形的诊断及微创治疗,对微创治疗后辅助手术方法修复复杂血管畸形颇有心得...
查看详细>>
熊学俊
外科(血管瘤/血管畸形)
从事血管瘤专业多年,现任合肥长兴康复医院血管瘤科主任,在临床各种血管瘤、脉管畸形疾病的诊治、手术操作娴熟。特别是在血管瘤与脉管畸形的诊断及微创治疗方面造诣较深,力争为每一位患者选择适合自身的诊疗方案...
熊学俊
血管瘤科主任
从事血管瘤专业多年,现任合肥长兴康复医院血管瘤科主任,在临床各种血管瘤、脉管畸形疾病的诊治、手术操作娴熟。特别是在血管瘤与脉管畸形的诊断及微创治疗方面造诣较深,力争为每一位患者选择适合自身的诊疗方案...
查看详细>>
龚洪涛
医学心理科
从事精神科临床工作十多年,熟悉内科、精神心理科常见疾病诊治,特别是对青少年心理障碍有较深的研究,并对精神心理疾病的辨证施治有着深入的研究和独到见解...
龚洪涛
精神康复科主任 / 资深心理治疗师
从事精神科临床工作十多年,熟悉内科、精神心理科常见疾病诊治,特别是对青少年心理障碍有较深的研究,并对精神心理疾病的辨证施治有着深入的研究和独到见解...
查看详细>>
曾海燕
外科(血管瘤/血管畸形)
从事我国婴幼儿血管瘤的工作多年,一直致力于血管瘤诊疗一线,深入研究血管瘤的发病机理及临床诊治,对血管瘤治疗方面有独到的见解。在诊治各种血管瘤疾病,擅长针对每个患者年龄、病因、病情制定个性化专属诊疗方案...
曾海燕
长兴康复医院院长
从事我国婴幼儿血管瘤的工作多年,一直致力于血管瘤诊疗一线,深入研究血管瘤的发病机理及临床诊治,对血管瘤治疗方面有独到的见解。在诊治各种血管瘤疾病,擅长针对每个患者年龄、病因、病情制定个性化专属诊疗方案...
查看详细>>
李凡
介入科 从事工作10年余, 熟练掌握导管室器械功能及用途,独立配台,巡台,熟练掌握无菌操作及各项注射技术,能有效的配合医生完成各项抢救工作。从事工作10年余,

庆娟
医学心理科
善于青少年心理问题的沟通工作、并制定饮食指导和健康宣教,有较强的语言沟通能力和协调能力

韦静
康复医学科
擅长:脑瘫康复,发育迟缓康复,运动骨折康复,罕见基因病康复,引导式教育。

李仁红
康复医学科
擅长儿童康复评估、抗痉挛体位的摆放、吞咽障碍康复训练、呼吸功能训练、清洁间隙导尿术、日常生活能力指导康复训练等。

刘月圆
护理部
从事护理工作10年,先后在内科、供应室、妇科工作,有丰富的理论知识和临床经验。

王晓明
成人康复
擅长中西医结合治疗脑梗、脑出血术后、偏瘫、脊柱损伤、截瘫、老年康复、疼痛康复、肩周炎、腰椎间盘突出症、植物人促醒脊髓损伤、周围神经损伤等疾病提供康复治疗。

封小平
检验科
从事检验工作十余年,熟练掌握临床检验相关操作、仪器维护保养及质量控制工作。

黄开宝
影像科
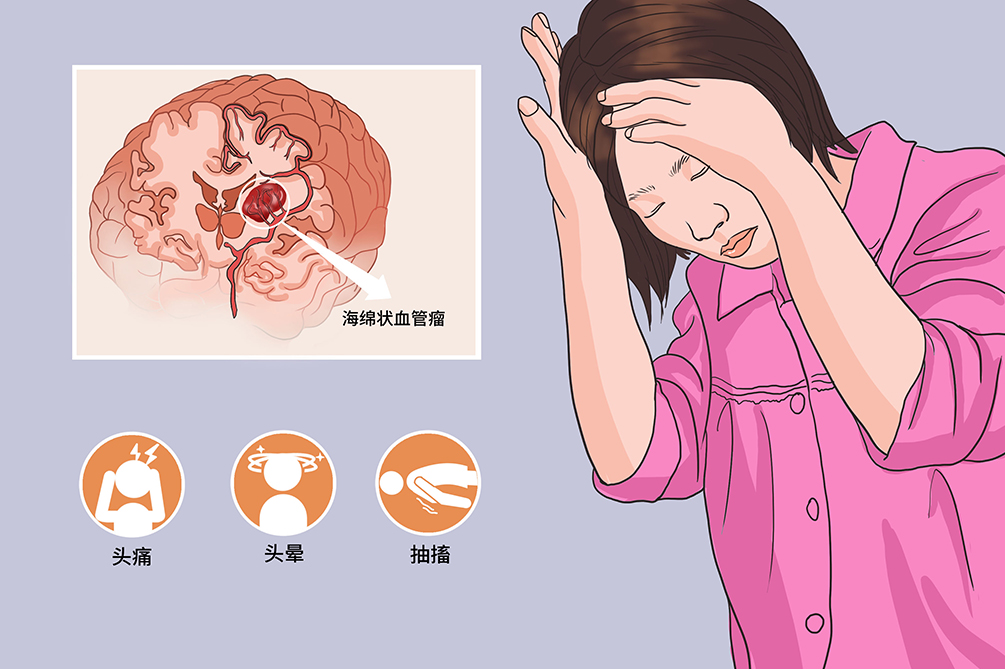
擅长放射医学影像DR、超声等医学影像的诊断,尤其是对各种常见的血管瘤、脉管畸形、海绵状血管瘤等多种疾病的检查识别。

钟世茹
医学心理科
⭐️擅长领域:儿童青少年心理健康、家庭亲子关系、人际交往困惑、压力和情绪调节、职场人际关系冲突、等从事团体咨询辅导。

童艳琼
医学心理科
从事精神心理医疗及教学工作30余年,熟练掌握失眠、抑郁症、精神分裂症、强迫症、神经衰弱等常见精神心理疾病的治疗方法,同时对多动症、抽动症等儿童行为障碍疾病的治疗也有深入的研究.....
童艳琼
副主任医师、教授、国家一级(高级)心理咨询师
从事精神心理医疗及教学工作30余年,熟练掌握失眠、抑郁症、精神分裂症、强迫症、神经衰弱等常见精神心理疾病的治疗方法,同时对多动症、抽动症等儿童行为障碍疾病的治疗也有深入的研究.....
查看详细>>
唐世早
肿瘤介入
肿瘤介入、腰椎间盘突出臭氧介入、下肢静脉曲张的泡沫硬化剂的介入、肝血管瘤等疾病的介入诊治造影技术。

王江恒
儿童康复
毕业于安徽医科大学,资深康复科医师,曾在康复专科医院从事临床康复学多年,在儿童发育行为与神经系统发育障碍性疾病领域颇有造诣,如幼儿吐字不清、发育迟缓、脑损伤儿的早期干、脑瘫、自闭症、多动症、抽动症、语言发音障碍、注高力障碍、学习因难、感觉统合失调的儿童等疾病经验丰富....
王江恒
资深康复医师(儿童康复)
毕业于安徽医科大学,资深康复科医师,曾在康复专科医院从事临床康复学多年,在儿童发育行为与神经系统发育障碍性疾病领域颇有造诣,如幼儿吐字不清、发育迟缓、脑损伤儿的早期干、脑瘫、自闭症、多动症、抽动症、语言发音障碍、注高力障碍、学习因难、感觉统合失调的儿童等疾病经验丰富....
查看详细>>
“三月春风送温暖 雷锋精神代代传”——合肥长兴康复医院义诊团队走进勤居苑社区

喜报!合肥长兴康复医院荣获“新站高新区三八红旗集体”称号

义诊进社区 关爱零距离——合肥长兴康复医院走进“勤居苑社区”

初心如磐 医路相伴丨合肥长兴康复医院党支部荣获市级“双比双争”先进社会组织党组织称号!

【合肥长兴康复医院】慰问康复患者,送上温暖与关怀
【义诊进社区】合肥长兴康复医院义诊团队走进“瑶海社区”

『长兴温度』别样小年!合肥长兴康复医院温情举措打造“长兴年味”

“重阳敬老情 义诊送健康”合肥长兴康复医院深入十里社区、勤居苑为老人看诊

全国助残日——合肥长兴康复医院残疾人文化进家庭“五个一”活动为群众提供医疗服务

义诊时老人突发癫痫!长兴康复医院的医生是这样做的

献爱心,送健康——合肥长兴康复医院

“迎八一,党 史学习教育”合肥长兴康复医院

【护士节】致全院护理工作者的慰问信

合肥长兴康复医院祝福所有教师—节日快乐

关爱女性健康 合肥长兴康复医院开展“两癌”筛查走进企业

“医心为民”合肥长兴康复医院党建联盟进社区第三弹!